

Research Projects
Understanding the nexus of nutrition, the microbiome, and virus susceptibility in the honey bee

Adam Dolezal and Irene Newton aim to understand how interactions between honey bees and their symbiont, Bombella apis, protect brood from environmental perturbations such as nutritional and viral stress. In prior research, the Newton lab has found that B. apis protects brood from nutritive stress. The Dolezal lab has found that both adult and larval nutrition impacts virus susceptibility in honey bee workers. Their project aims to connect these two lines of inquiry, identifying links between B. apis colonization, nutritional supplementation, and virus susceptibility in both honey bee workers and queens. Currently, the Dolezal lab is further exploring the dimensions of the mutualistic effects of B. apis in larval honey bees by measuring the combined potential of their nutritional buffering and xenobiotic detoxification abilities. When raised naturally inside a hive, honey bee larvae are exposed to an enormous variety of outside influences, ranging from pesticides accumulated in the wax cells to diseases spread by parasitic mites. Manipulating the larvae in vitro using artificial diets and sterile well plates allows for a much finer level of control over their nutritional, chemical, and microbial environments and can better tease apart the interactions between larval microbiomes and their influence on honey bee health.
Establishing a GEMS culture collection

Very few systems of study in biology include molecular level understanding of coevolution among multiple interactors since most studies still focus on pairwise interactions. An important aspect of GEMS is basic discovery – capturing natural variation in a diversity of microbial species within our single focal environment. Toward this end, Rachel Whitaker, Bill Metcalf, and many others are leading an effort that includes both targeted and general culturing strategies to capture rhizobia, bombella, pseudomonads, streptomyces, and fungal species plus many new bacteria from field sites at the W.K. Kellogg Biological Station. Culturing will be paired with culture-independent sampling of soil, leaf, and other environments and expanded in future years to capture microbial viruses and other elements.
Mutualism response to stress

Symbiosis with rhizobial bacteria is a strategy legumes employ to meet one stress - the challenge of nitrogen limitation. In previous work, this research team has documented a degradation of this symbiosis in the long-term nitrogen fertilization plots at the W. K. Kellogg Biological Station (KBS), where rhizobia from high N plots were less beneficial for host plants. However, nitrogen availability is only one dimension of the environmental context in which the clover-rhizobium symbiosis exists. How does a system so sensitively tuned to one stressor (e.g., nitrogen) respond when confronted with another major environmental stressor? Does the partnership collapse under the pressure of multiple and potentially conflicting constraints, or do the eco-evolutionary feedbacks inherent in the symbiosis contribute to rapid adaptation? To what extent are relationships with other partners (e.g., "symbionts of symbionts" and members of the plant microbiome) involved in the response? Tony Yannarell, Jen Lau, and others are investigating the constraints that may limit adaptation to multiple stressors (nitrogen, drought) using the clover-rhizobium symbionts from KBS plots. They hypothesize that relationships with a broad array of partners in the microbiome can help the clover-rhizobium symbioses adapt to multiple environmental stressors.
Mutualism feedbacks on plant-insect interactions

The clover-rhizobium and bee-virus systems are linked through plant traits that have important implications for bee biology. Furthermore, the clover-rhizobium system is also important to multiple insect herbivores which depend on the nutrient-rich tissue of legume plants. Therefore, we hypothesize that the evolution of less cooperative rhizobia in response to long-term nitrogen (N)-addition will affect interactions with pollinators and herbivores because of changes in floral and other plant traits. Adam Dolezal, Jen Lau, Alex Harmon-Threatt, and others will test these hypotheses by quantifying the effects of both N and rhizobium evolution on plant traits and investigating how these traits affect bee visitation, behavior, and physiology, as well as herbivore survival and development. This starting point will facilitate future work that aims to identify how observed changes in plant traits affect bee immunity and health, as well as how rhizobium evolutionary effects on bee visitation feeds back to affect plant demography and might contribute to observed legume declines in high N environments in the field.
Recombination, HGT, and natural selection in bacterial evolutionary responses
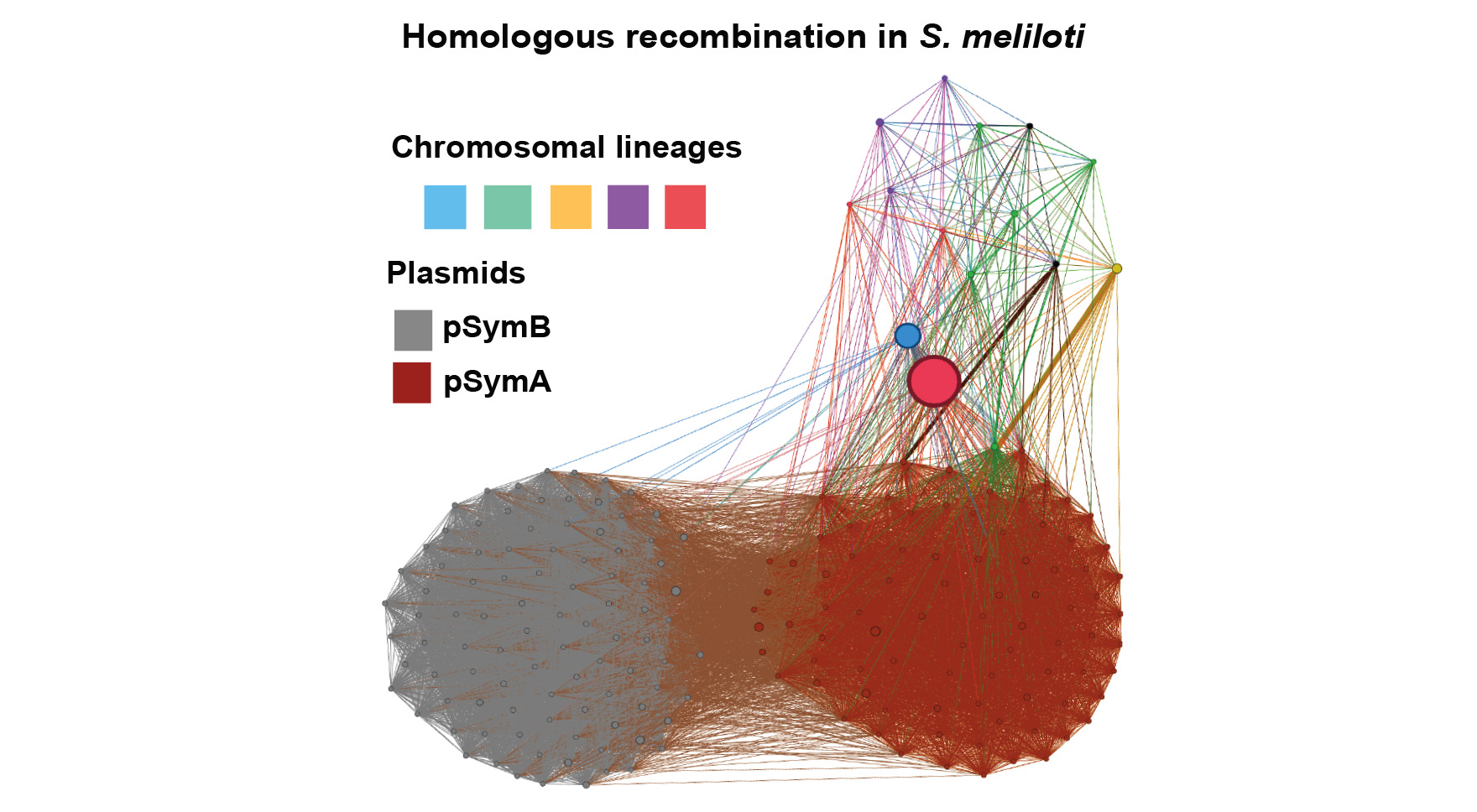
It is clear that horizontal gene transfer (HGT), acting at various phylogenetic scales, is responsible for major evolutionary innovations in microbes. Detecting HGT, particularly at local/population genomic scales, is a difficult and potentially computationally- and statistically-challenging problem. We have yet to settle on a set of comprehensive and generalizable methods for detecting recombination and HGT in bacterial genomes so that these patterns can be combined with signatures of natural selection to address evolutionary responses in ecologically relevant traits in natural bacterial populations. In Rhizobium symbionts of legumes, for example, the region of highest differentiation between control and N-evolved strains (which have become less beneficial for plant hosts) appears to have been transmitted across a diverse set of chromosomal lineages. Also, another legume symbiont, Sinorhizobium meliloti, exhibits an intricate pattern of gene transference among chromosomal lineages and replicons (chromosome and megaplasmids), which involves homologous recombination and gene loss. However, addressing these problems with rigor will require additional focused work and novel methods. Using existing datasets consisting of hundreds of rhizobial strains, GEMS postdoc Mario Ceron Romero is working with Katy Heath, Rachel Whitaker, and Tandy Warnow to test existing methodologies and develop novel pipelines to address these questions, which can be applied to Bombella and Pseudomonas populations and beyond as part of ongoing research in the Institute.
Eco-evolutionary forces maintaining genetic variation in Pseudomonas syringae
Tremendous diversity, both in terms of core genomes and effector repertoires, can be maintained in populations of the plant pathogen, Pseudomonas syringae. The labs of Joy Bergelson, Mercedes Pascual and Rachel Whitaker have come together to combine coevolutionary theory with empirical studies of plant – bacterial interactions to understand the eco-evolutionary forces maintaining this genetic variation. The team aims to develop theory to explore how microbes’ non-core genomic elements, particularly virulence factors that are secreted by pathogen isolates, shape stable persistence of genomic diversity among pathogens. These non-core genetic elements are good candidates for driving eco-evolutionary dynamics because they sit at the frontline of the interaction between host and pathogen and are specific in terms of which plant genotypes elicit a response. If host resistance responses have appropriate levels of specificity, then these non-core genetic elements could drive the maintenance of pathogen diversity. We are generating data to determine the degree of specificity in resistance responses to inform our models and are examining genomic patterns of association among effectors as an indicator of eco-evolutionary dynamics. While our focus is currently on the host plant, Arabidopsis thaliana, we are also generating data for P. syringae from crop plants. We plan to extend our model to include lateral gene transfer (LGT) and mutational processes in the generation of dynamics. Given LGT, these non-core elements can be viewed as “embedded” and symbiotic to the microbial pathogen genome.
Coevolutionary interactions between clover hosts and rhizobium symbionts

Nitrogen (N) addition is predicted to destabilize the legume-rhizobium mutualism. Lau and Heath have previously found data to support this theoretical prediction for the rhizobium partner. The potential role of evolution in the legume host in response to N-addition, or how N-addition will alter the coevolutionary feedbacks between leguminous hosts and their rhizobium symbionts, remain unaddressed. Jen Lau together with Katy Heath, Joy Bergelson, Carla Caceres and others will use approaches from quantitative genetics to investigate the potential for host evolutionary responses to N and to evolutionary changes in their rhizobium partners. Their initial project will identify the extent of plant genetic variation in traits underlying symbiotic interactions with rhizobia and predict evolutionary responses of plants to both N and rhizobium genetic variation. The goal is to begin to better understand the responses of the plant component of this series of nested mutualisms and the potential for coevolution between the plant and rhizobium partners. These results and generated plant lineages will lead to further experiments assessing how plant evolution feeds back to influence interactions with other community members and rhizobium evolution.
Evolutionary ecology of symbiosis using microbial mark-recapture
Over the past few decades, experimental procedures have been developed and refined for the study of microbial evolution under controlled laboratory conditions. Whether such information from test-tube studies pertains to the evolution of microorganisms and their hosts in nature is uncertain owing to the challenges associated with tracking genotypes and populations in the wild. One promising and recent development involves the insertion of barcodes into the genomes of microorganisms, which can then be retrieved through quantitative sequencing technologies. GEMS postdoc John McMullen has developed a population genomics pipeline for molecular barcoding of closely related strains with the goal of quantifying fitness costs associated with the varying lifestyles of microbes. McMullen, together with Jay Lennon, Katy Heath, Jen Lau, Cari Vanderpool, and Rachel Whitaker, is using this strategy to study the evolutionary ecology of Rhizobium leguminosarum, both during its interactions with clover hosts and in the soil environment. In particular, they are focusing on bottom-up and top-down pressures of nitrogen addition and phage parasitism, respectively. The project goal is to quantitatively track genotypes (i.e., phage defense systems) in nested symbionts to understand trade-offs and evolutionary constraints on mutualisms involving plants, microbes, and mobile genetic elements. The proposed work addresses long-standing limitations that preclude a full understanding of the legume-rhizobium symbiosis, including challenges associated with quantifying fitness and evolutionary trajectories of bacteria in soil and plant life-stages in the face of disruptive effects. Molecular-based mark-recapture approaches will synergize with other GEMS-related research that seek to quantitatively understand the evolution of microbial populations (e.g., Pseudomonas, Streptomycetes, Bombella) embedded in complex communities.
Symbiosis genes, HGT, and mutualistic partner quality

Previous genomic work comparing the more beneficial rhizobium symbionts from ambient nitrogen environments to the less beneficial rhizobia from high nitrogen environments showed that genetic variation at the symbiosis gene region of the pSym symbiosis plasmid differentiated high and low-quality populations. The specific loci in this region are canonical symbiosis genes, including regulators and enzymes required for nitrogen fixation, but the effects of specific mutations on symbiosis phenotypes have not been determined, despite the ability to manipulate Rhizobium in the laboratory. In addition, the phylogenetic evidence to date suggests HGT of this region of the plasmid, across diverse chromosomal backgrounds, making this an even more interesting candidate genetic region for functional validation to link selection and HGT. Cari Vanderpool, Katy Heath, Rachel Whitaker, Amy Marshall-Colon and others will integrate molecular genetics with quantitative genetics and evolutionary ecology to address this current gap in knowledge. Combining additional analyses of sequenced genomes with exploration of transcriptional profiles and targeted genetic manipulations will allow us to address evolutionary responses of mutualisms at a mechanistic level, integrating molecular processes with ecological processes in this system for the first time.
Defining the functional diversity and evolution of phage-associated endosymbiont toxins
Bacterial endosymbionts hijack host cell biology by secreting effectors that mimic eukaryotic protein function. These effectors encode domains that are homologous to eukaryotic toxins and tend to cluster inside phage genomes that infect endosymbionts. Phage-associated toxins can have dramatic impacts on host cell biology. For example, expression of phage-associated toxins underlies cytoplasmic incompatibility (CI) between males and females infected with the maternally transmitted endosymbiont Wolbachia in Drosophila and mosquitoes. But, expression of non-phage-associated toxins does not appear to affect CI. It remains unclear, therefore, whether association with the phage genome itself impacts the function and evolution of endosymbiont toxins. Tandy Warnow and Irene Newton address this question by comparing the functional diversity and molecular evolution of phage- versus non-phage-associated toxins from Wolbachia. Over the last ten years, hundreds of diverse endosymbiont and associated phage genomes have been sequenced, but the impact of this diversity on the coevolution of host-symbiont interactions remains unclear. This project aims to capture much of the functional arms race between host, symbiont, and phage by characterizing the diversity and evolution of horizontally transferred endosymbiont toxins.
Fungal contributions to GEMS systems

Fungi are underappreciated components of microbial communities and can contribute to community assembly dynamics through microbe-microbe and host-microbe interactions. Specifically, Aspergillus species are ubiquitous environmental fungi that can play significant roles in plant, bee, and soil health. These species also unify the GEMS systems and can be potentially trafficked between them. However, little is known about the phenotypic and genetic diversity of Aspergillus strains in any of the GEMS systems. For example, previously, the Berenbaum and Dolezal labs have shown that a specific Aspergillus flavus strain, isolated from bee bread, can detoxify pesticides, providing a benefit to the bee. However, a different isolate of Aspergillus flavus, cultivated from a dead honey bee by the Newton lab, is a potent pathogen of bee brood and is inhibited by the B. apis antifungal metabolite. These disparate results lead to questions such as: What is the genetic diversity of Aspergillus strains across these environments? How does this diversity relate to the ability of these organisms to interact with resident microbes and the bee colony? Is there environmental filtering that occurs as the bees traffic in these environmental microbes from gathered pollen? What is the spillover effect on bee health? May Berenbaum, Irene Newton, Adam Dolezal, and Tony Yannarell focus on the bee system as preliminary data from the group suggests Aspergillus may play significant roles in the health of the bee colony. However, because the colony is an open system, the project and framework easily extend to other GEMS systems, with the potential for this project to expand to the soil, and plant interactions in subsequent years.
Molecular characterization of genome conformation in rhizobium symbionts of clover
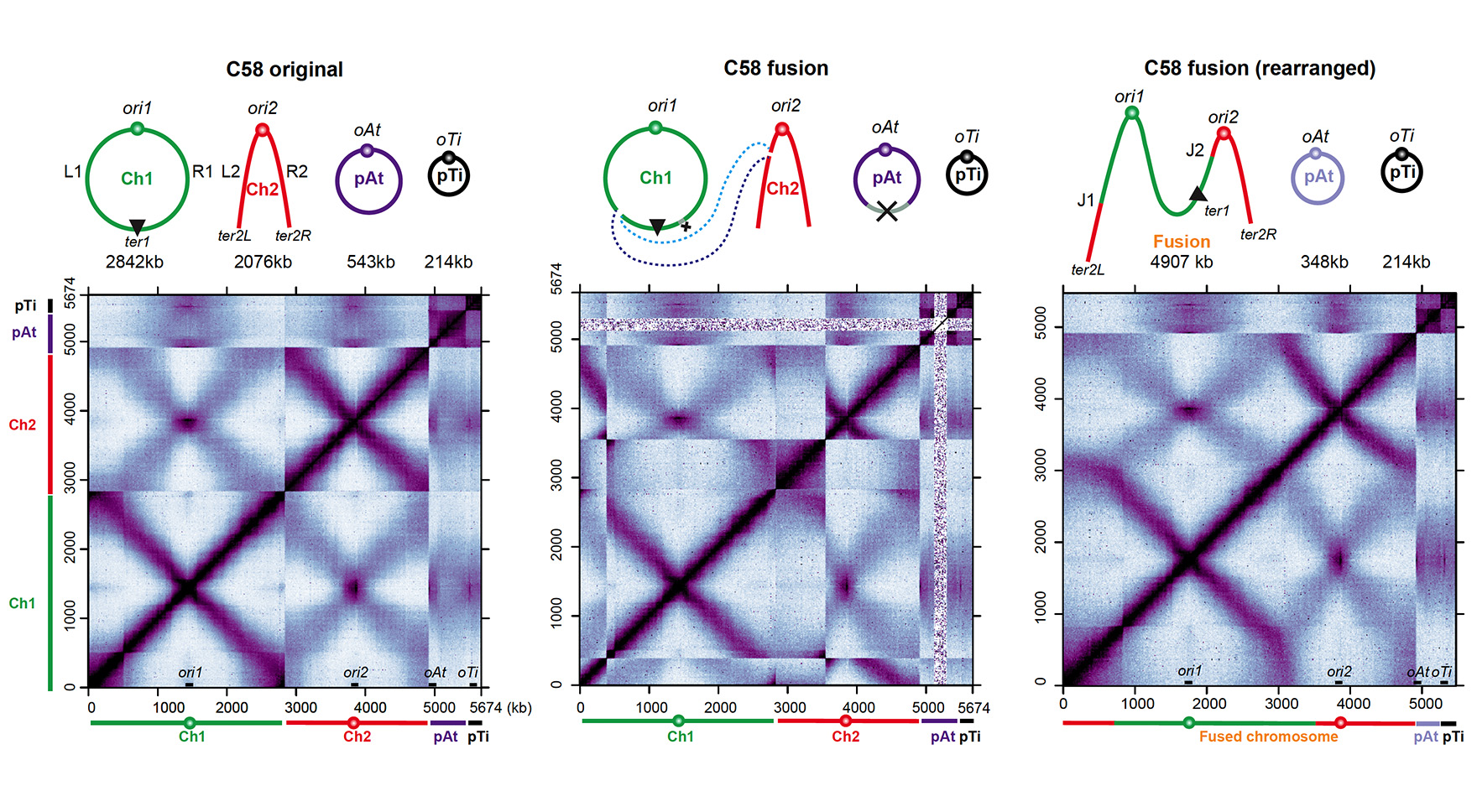
Mutually beneficial species interactions are universal in natural ecosystems, including agriculture and the human microbiome. Mutualism enables the exchange of resources and, combined with a changing environment, is hypothesized to drive inter-species co-evolution. However, the molecular mechanisms that link mutualism to evolution are lacking. Xindan Wang, in collaboration with Katy Heath and Cari Vanderpool, will utilize the mutualism of legume and nitrogen-fixing rhizobia to understand the molecular basis for evolutionary differentiation through the lens of genome architecture and gene expression. The project aims to investigate the genome architecture of rhizobia variants that have differential symbiosis ability with clover by performing genome-wide chromosome conformation capture assays (Hi-C) on rhizobia strains with different genome rearrangements. Furthermore, they strive to understand the alteration of gene expression in the evolved rhizobia strains via RNA-seq experiments in the strains with differential symbiotic abilities. This collaborative project will address the Rules of Engagement theme in GEMS and result in the development of molecular tools to study genome conformation and gene expression that can be used in other model systems.
Plasmid population biology across environments in Rhizobium leguminosarum

The resource mutualism between legumes and rhizobia is an emerging model for how nested symbioses evolve. Nitrogen (N) fixation is a plasmid-encoded function in Rhizobium leguminosarum; thus, a greater understanding of the dynamics of plasmids in natural populations is critical for understanding how plasmid costs and benefits might shift under different selective environments and drive mutualism evolution. Previous work has shown that long-term N fertilization has resulted in fewer hosts, fewer symbiotic rhizobia, and lower-quality rhizobium partners for plant hosts. GEMS graduate student Sierra Bedwell, working with Katy Heath and Rachel Whitaker, hypothesizes that the abundance of plasmids (including pSym which carries the canonical symbiosis genes) might also change across environments and serve as a driver of mutualism decline. To test this, the team must consider strains in the soil in addition to nodule isolates, which are sampled in symbiosis with plants. The project aims to track plasmid dynamics in natural populations and widen the sample of Rhizobium strains from the soil towards better understanding the mechanisms of mutualism decline in nature.
Untangling multiple ecological drivers of rhizobium evolution under nitrogen enrichment

Nitrogen (N) enrichment causes rhizobia to evolve reduced mutualist quality, but N also decreases light availability and legume abundance. Therefore, it is unclear whether rhizobium evolution is due to the direct effects of N, indirect effects that arise over longer timescales as N-addition affects plant communities (light or legume abundance), or their interaction. GEMS graduate student Mackenzie Caple, working with Jen Lau and Katy Heath, has inoculated greenhouse mesocosms with soil from 30-year-old Kellogg Biological Station Long-Term Ecological Research (KBS LTER) N-addition or control plots and factorially manipulated light, N, and legume density to investigate the interactive roles of these factors on rhizobium evolution. They are also investigating whether rhizobium quality recovers following cessation of N enrichment. These experiments allow for the identification of selective agents responsible for observed declines in mutualism, comparison of genetic changes after selection in different environments, and investigation of the reproducibility of evolution.
Multi-scale structure of natural variation in Streptomyces in rhizosphere of long-term nitrogen addition experiment

Microbial populations are vital to interactions in the ecosystem; however, it is difficult to predict how microbial communities and populations vary in response to environmental change.
We are investigating changes in the community and population dynamics in the genus Streptomyces within the rhizosphere under long-term nitrogen addition as a model of how environmental changes impact microbial communities across scales. These long-term nitrogen addition sites (W.K. Kellogg Biological Station Long-term Ecological Research) provide a context in which to observe microbial evolution in the rhizosphere across scales, in response to nitrogen addition at levels comparable to agricultural fertilization. GEMS graduate student Isabelle Lakis will work with Rachel Whitaker, Katy Heath, and other members of the GEMS community to investigate the population structure and dynamics of environmental Streptomyces across scales. They hypothesize that long-term nitrogen deposition and plant association both diverge Streptomyces communities and populations and cause variation in Streptomyces pangenomic elements, including natural products.
Influence of the mobilome on the honey bee microbiome via longitudinal metagenomics and reverse ecology

The honey bee gut microbiome is influenced by a mixture of ecological factors, such as invasion by external strains or parasites, and by evolutionary factors, such as drift, selection, and horizontal gene transfer (HGT). Despite long-standing theoretical debate, it is not well known how short-term evolution of host-associated microbial species feeds back and affects the interactions within the local community – especially in a complex microbial environment such as the honey bee gut microbiome. Further, it is not well-known how, and to what frequency, interactions between mobile genes and their hosts affect the evolution and composition of honey bee microbial communities. To address the influence of mobile gene ecology and evolution in the honey bee microbiome, GEMS graduate student Chris Robinson, in collaboration with Irene Newton, Adam Dolezal, Rachel Whitaker, and Mercedes Pascual, will conduct longitudinal analysis of apiaries at IU and UIUC.
Effects of diet and environmental stressors on the microbiota mobilome from honey bee queens

High-throughput sequencing has changed the way we can study the complexity of microbiomes, going beyond just describing the taxonomic and genetic diversity of symbionts. An important part of any microbial community is the mobilome, the set of mobile genetic elements (MGEs). For instance, plasmids can carry important resistance genes or virulence factors, transposable elements can modify gene function and regulation, and phages can kill the host or integrate its DNA. Thus, studying MGEs is essential for a better understanding of the microbiome assembly and function. The microbiome of worker honey bees and its beneficial role for host health has been intensively studied, but the impact or role of the mobilome in shaping the microbiome or the bee’s health is still unclear. This knowledge gap is even more pronounced for bee queens, the sole reproductive unit of the colony. Adam Dolezal and GEMS postdoc Lílian Caesar (Newton Lab) propose to integrate research systems independently developed in the Dolezal and Newton labs to test the effects of diet and xenobiotics on queen fitness and then characterize the microbiome and mobilome.
Effect of Long-term Fertilization on the Evolution of Legume-Rhizobia Symbiosis

Long-term N addition can cause disruption of the legume-rhizobium mutualism, possibly due to the degraded soil diazotroph pool, the weakened plant-microbe feedback pathways, disrupted plant selective mechanisms, and the decreased propensity to form nodules. Since the legume-rhizobium symbioses is the main contributor of natural fixed nitrogen to terrestrial ecosystems, a thorough characterization of diazotroph community structure and legume symbioses efficiency under long-term managements of fertilization is critical to benefit agricultural productivity and sustainability. The Morrow Plots is an agricultural experiment that contains unfertilized plots as well as plots that receive organic and inorganic fertilizer, as well as two rotations that include legumes (soybean and alfalfa). The 147-year duration of this experiment makes it uniquely suited for characterizing the evolutionary consequences of long-term fertilization on legume-rhizobium symbioses in this project led by Angela Kent and graduate student Danyang Duan, in collaboration with Tony Yannarell.
High Fitness Burden of Virulence Drives Genome Evolution in a Facultative Plant Pathogen
Facultative pathogens, by definition, can persist away from their hosts and thus their capacity to cause disease is greatly impacted by the stability of virulence functions during host association and in their host-independent reservoir(s). Agrobacterium tumefaciens is a facultative pathogen that causes crown gall disease on plants through horizontal gene transfer (HGT) directed by virulence functions that are predominantly encoded on the tumor-inducing (Ti) conjugative plasmid. The Ti plasmid and another conjugative mega-plasmid known as the At plasmid impose significant carriage costs on A. tumefaciens that are exacerbated by nutritional stress. Virulence (vir) gene expression is tightly regulated by plant-released signals. Plant tissues expressing A. tumefaciens transgenes excrete semi-private nutrients called opines that support agrobacterial rhizosphere populations proximal to the infection site. Our studies in laboratory culture have revealed that Ti plasmid fitness costs are very high when virulence is activated, and mutations disabling vir genes, as well as Ti plasmid curing, are common after cultivation under vir-inducing conditions. However, whole genome sequencing of A. tumefaciens populations infecting plants suggests that the bulk of the population does not experience elevated plasmid loss or mutation of virulence functions. Clay Fuqua and graduate student Ian Reynolds hypothesize that there is a small infecting population in intimate physical association with infection sites. This spatially distinct subpopulation drives the HGT process and incurs the fitness burden but is greatly outnumbered by the bystander population that can benefit from the result of pathogenesis and do not experience its cost. A genetic approach is being developed to test this hypothesis and interrogate specifically the subpopulation that is activated for virulence and cross-kingdom horizontal gene transfer.
Multipartite genome evolution in E. coli
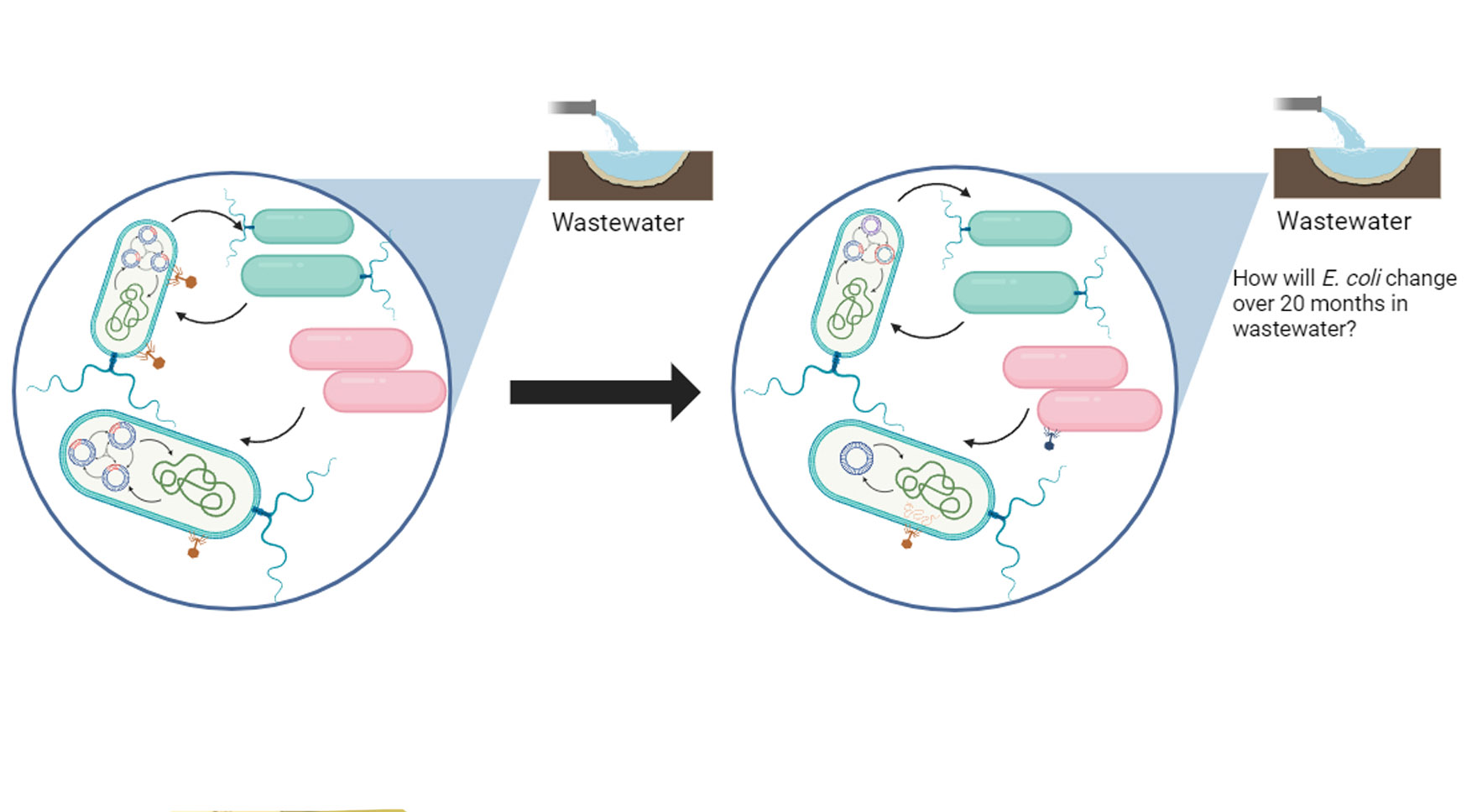
E. coli is perhaps the most studied microorganism, with nearly 100,000 publicly available genome sequences. Analysis of these genomes shows excellent diversity within this species structured into very different sequence types. Most sequence types have global distributions but different associations with habitats and hosts. Comparisons seeking to understand the movement of antibiotic resistance in the local One Health context of clinical, animal, and environmental distributions show strong evidence for clonal (whole genome) human-to-human transmission but very little evidence for sharing E. coli clones between human, animal, and ecological habitats. These One Health studies do, however, uncover sporadic sharing of identical antibiotic and virulence genes and very rarely identical plasmids. This suggests that the mobility of both genes and MGEs (mostly plasmids) at the nested sub-genomic scale is unlinked from host chromosomes but likely driven by cell type abundance in different habitats. The next horizon of these studies is to establish predictive models for gene flow, including the biological barriers to transmission and coinfection of MGEs that might promote the transmission of plasmid-carrying genes. Graduate student Isaiah Goertz and Rachel Whitaker, in collaboration with Helen Nguyen at UIUC, propose a large genomic study to determine the plasmid distributions and colocalizations within and between E. coli host cells from populations collected longitudinally from wastewater samples over 20 months for COVID surveillance.
Viral infection changes the outcome of bacterial evolution

A large majority of all sequenced bacterial isolates are latently infected by viruses; however, our understanding of how the viruses affect the evolutionary trajectories of their partners is understudied, and the benefit to the viruses within this context is often considered only from a bacterial perspective. Graduate student Laura Suttenfield and Rachel Whitaker, in collaboration with James Slauch and Zoi Rapti at UIUC, propose to assess how viral fitness evolves in a context where vertical transmission is enforced by a lack of susceptible hosts. They hypothesize that selection for viral vertical transmission results in lowered spontaneous induction of the virus and adaptation of the hosts to their virus before the environment.
Closed genomes of Rhizobia populations for evolutionary analysis
Molecular understanding of microbial genomes shows that complete genomes will be required for eco-evolutionary analyses that include variable elements of microbial genomes, including viruses, plasmids, defense islands, CRISPRs, and large indels. The polished genomes will integrate evolutionary dynamics with genome architecture and mutation and recombination mechanisms and HGT balanced with selection. Rachel Whitaker and the GEMS core team propose long-read sequencing of 65 strains of Rhizobia isolated from 2008 that form the basis of GEMS mutualism breakdown with N addition. An additional 60 strains isolated in 2018 will also be added for full analysis as well as a growing set of strains (up to 50) from leaves and soil in N addition and control plots isolated by the GEMS culture club.
From sequence to structure workflow for analysis of CRISPR-Cas immunity dynamics in microbial populations
Predictive models for the epidemiology of infection by genetic elements of microbes are necessary to address both their ecological and evolutionary impacts. Metrics have been developed that describe the diversity of the CRISPR spacers and their outcome in simple microbial communities; however, their comparative application to empirical data sets is limited. Rachel Whitaker, Irene Newton and colleagues will develop an accessible genomics tool for analysis of CRISPR population diversity in populations of bacterial genomes.